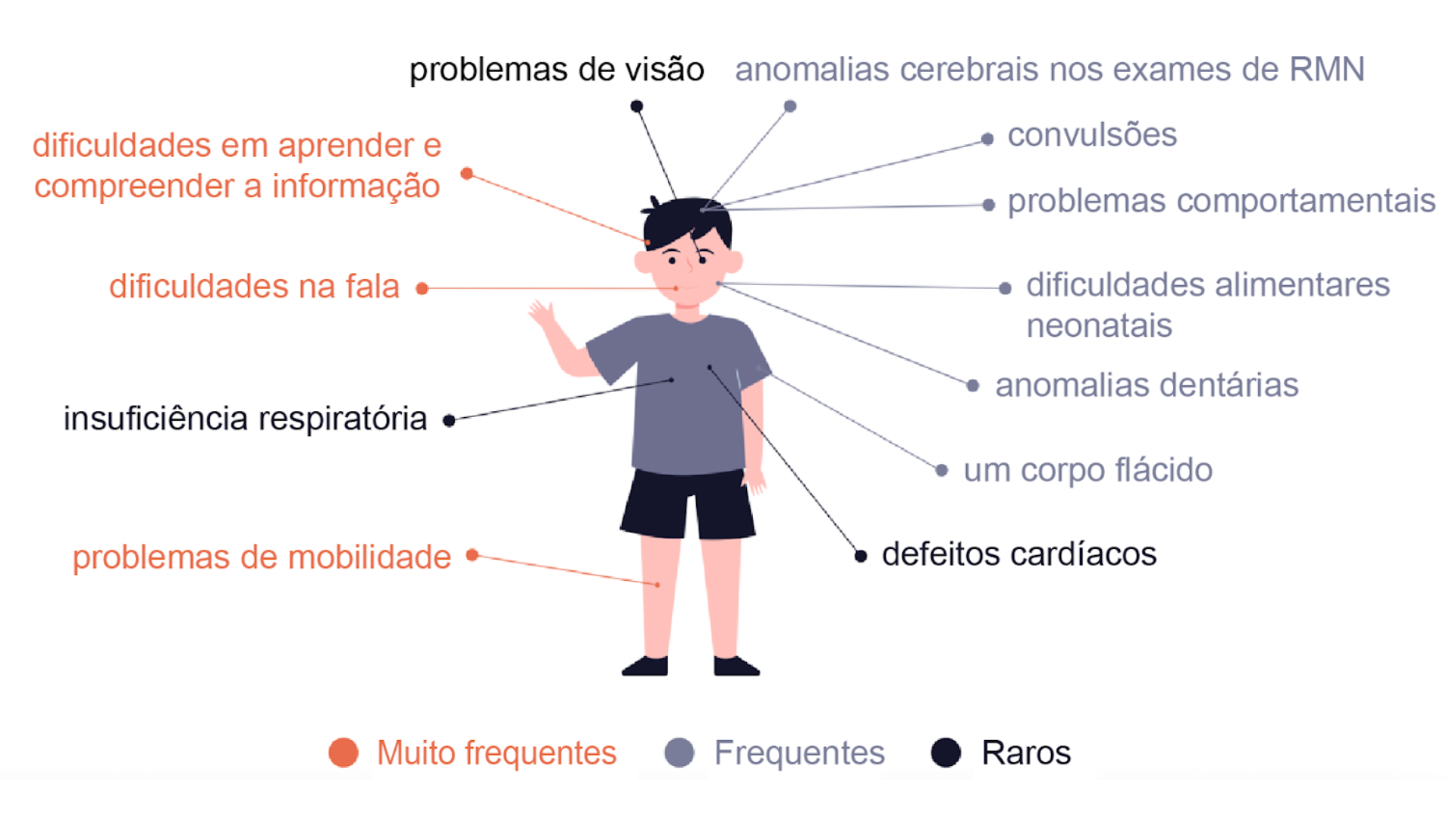
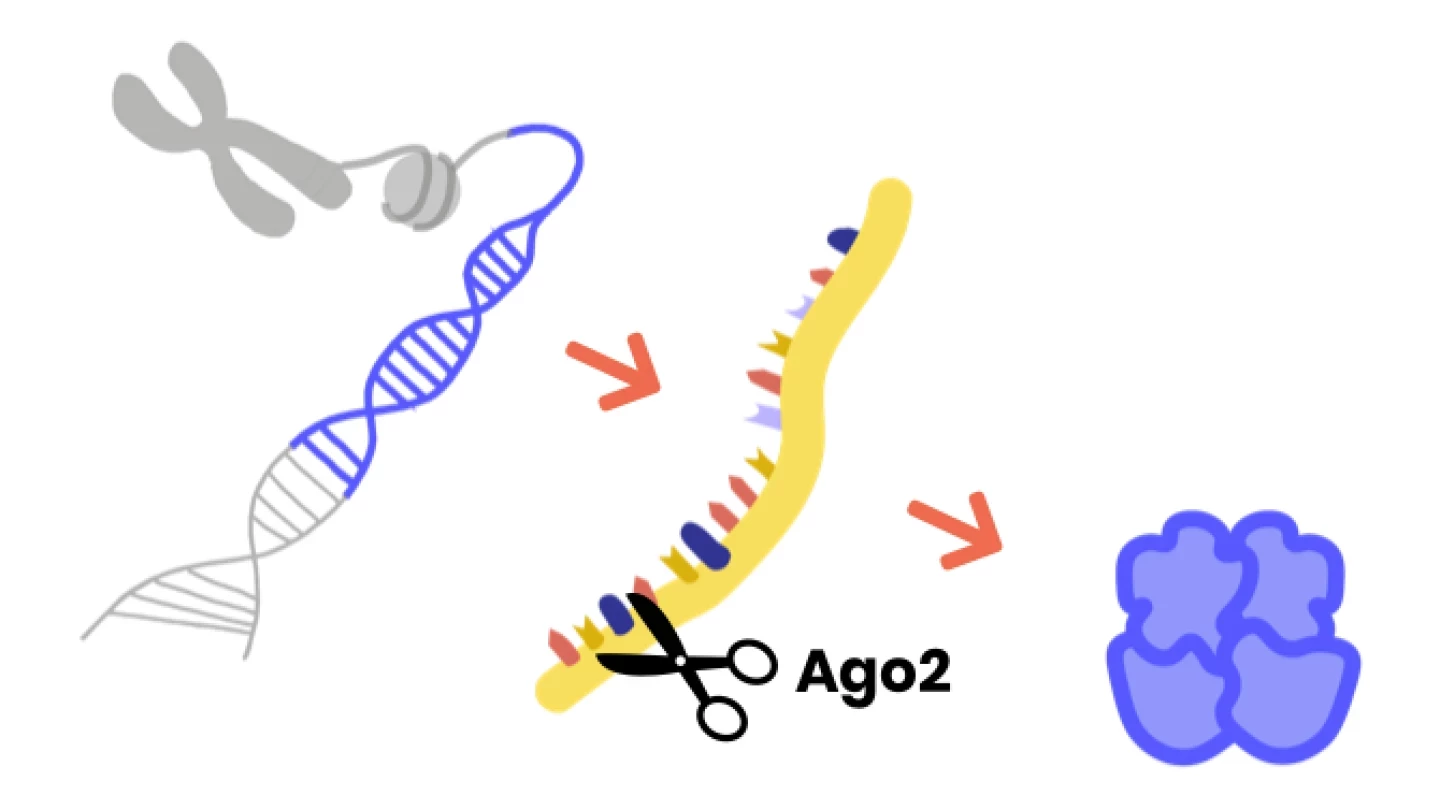
As síndromes de Argonaute são transtornos de neurodesenvolvimento raros, caracterizadas por atraso no desenvolvimento motor, convulsões, problemas na fala e compreensão, e défice cognitivo.
Estão associadas a alterações que causam a doença nos genes de Argonaute, especificamente Argonaute1 (AGO1) e Argonaute2 (AGO2).
O Prof. Piton descreveu a síndrome associada ao AGO1 em 2021. O Prof. Lessel e o Prof. Kreienkamp descobriram a síndrome associada ao AGO2 e descreveram-na, pela primeira vez, em novembro de 2020. Esta última foi designada de “síndrome de Lessel-Kreienkamp”, no início de 2021 (OMIM).