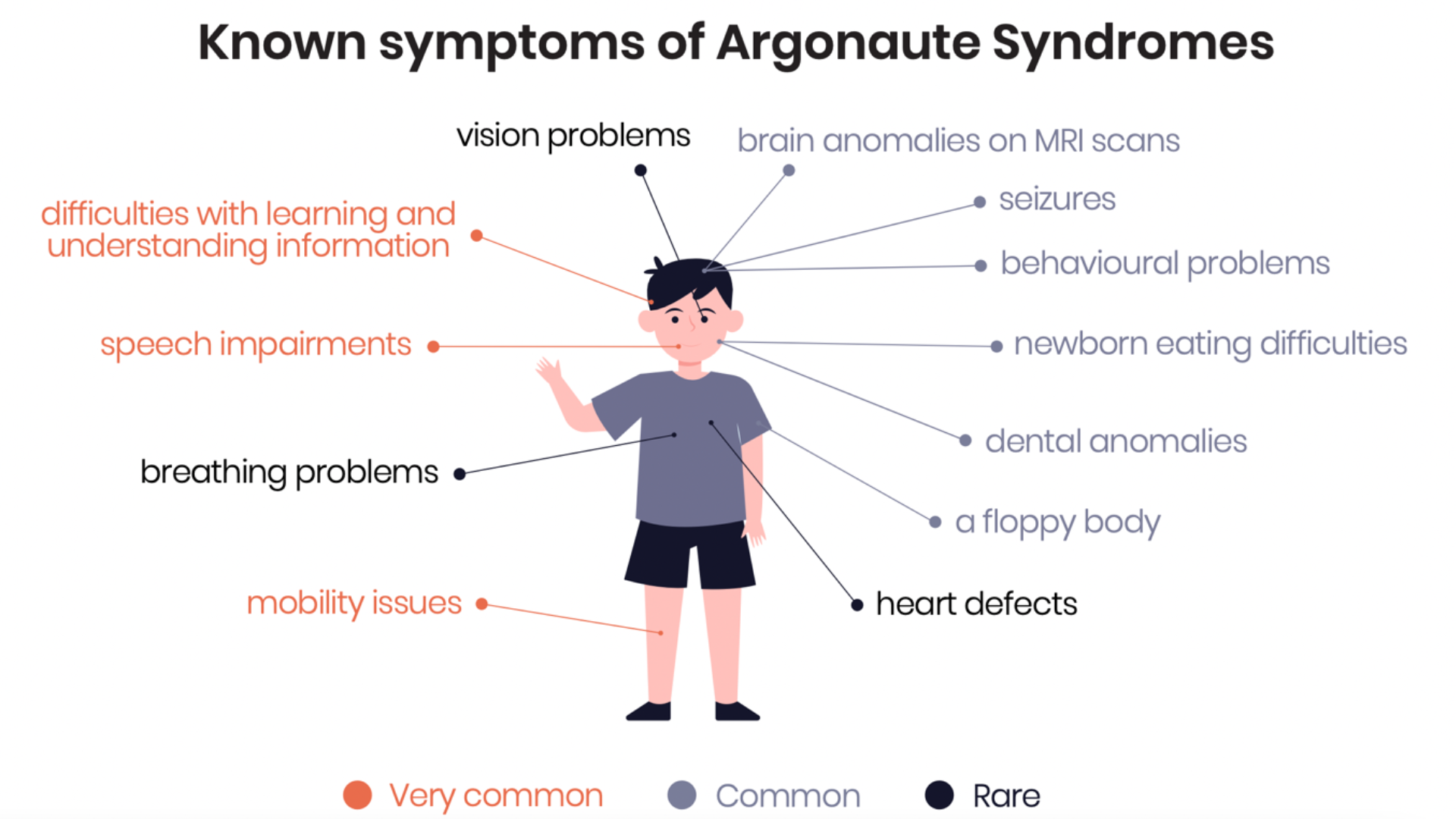
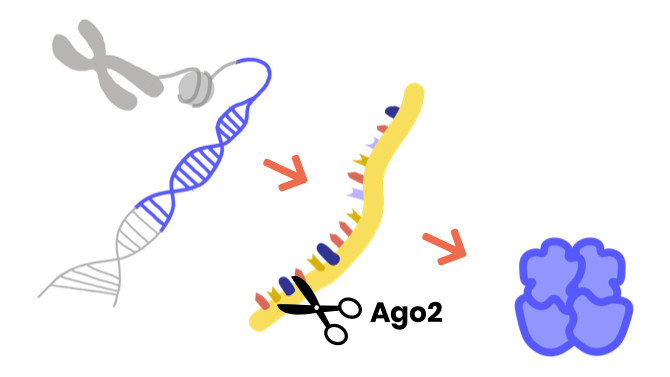
Argonaute syndromes are rare neurodevelopmental disorders characterized by delayed motor development, seizures, problems speaking and understanding, and cognitive impairment.
They are associated with disease-causing changes in the Argonaute genes, specifically Argonaute1 (AGO1) and Argonaute2 (AGO2).
Prof. Piton described the AGO1-related syndrome in 2021. Prof. Lessel and Prof. Kreienkamp discovered the AGO2-related syndrome and first described it in November 2020. The latter was named "Lessel-Kreienkamp syndrome" in early 2021 (OMIM).